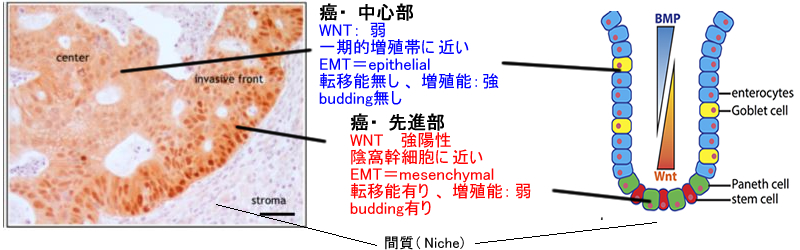
論文の主旨は「原発巣と転移巣のゲノムの比較」をし、超早期転移なら、両者の差は無く、晩期転移なら差がある(転移巣に新たなドライバー変異が見つかる)はずだが(下図)・・・結果は転移巣で新たな変異(metastasis-private mutations)は少なく、前者(超早期転移)の可能性が支持された、という内容である。
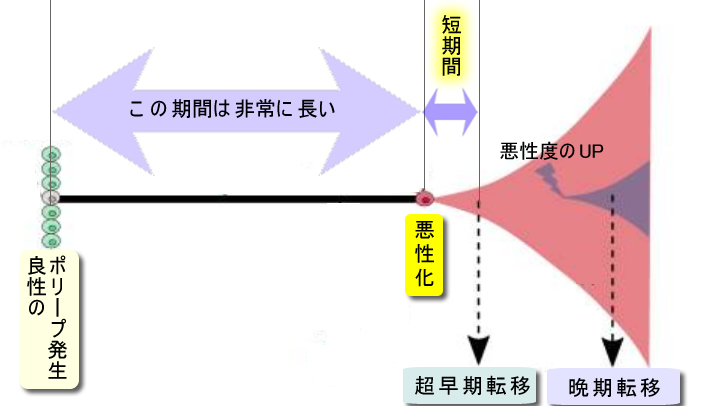
超早期転移の問題は換言するなら「Canonical Driversだけで転移が起こるか否か」という問題になる
「Canonical Drivers だけで転移が起きる」という報告として「APC, EGFR, TP53 、TGFβ4の4つの変異を導入した正常な人の腸細胞は転移するという2017年文献がある。
一般化するなら
転移の律速段階である以下の(1)~(6)がCanonical Driversだけで起こることが確認されれば「超早期転移の分子的証明」になる。
| (1)細胞-細胞結合の消滅(EMTマスター遺伝子発現) (2)運動能の亢進(Rho,RACによるアクチンの再構築など) (3)細胞外マトリックス分解酵素の増加) (4)Angiogenesis(新生血管は細胞密着がゆるいので、血管への侵入は障壁ではない) (5)幹細胞化 (6)免疫回避 (PD-L1の発現) |